
Por Lourdes Morro Felipe*

Muchas veces compramos medicamentos con nombres que a veces no
comprendemos. Uno de estos medicamentos, y uno de los que más se habla
en los últimos tiempos, es el captopril, un inhibidor de la enzima de
conversión de la angiotensina (IECA), que consumen muchas personas con
hipertensión.
El origen de la hipertensión está vinculado con el sistema renina-angiotensina, conocido desde finales del siglo XIX, cuando Tiergerstedt y Bergman demuestran la existencia de una sustancia presora presente en los extractos crudos de riñón. Aproximadamente 40 años después, dos grupos independientes de investigadores descubren que esta sustancia presora es un péptido formado por la acción catalítica de la renina.
El origen de la hipertensión está vinculado con el sistema renina-angiotensina, conocido desde finales del siglo XIX, cuando Tiergerstedt y Bergman demuestran la existencia de una sustancia presora presente en los extractos crudos de riñón. Aproximadamente 40 años después, dos grupos independientes de investigadores descubren que esta sustancia presora es un péptido formado por la acción catalítica de la renina.
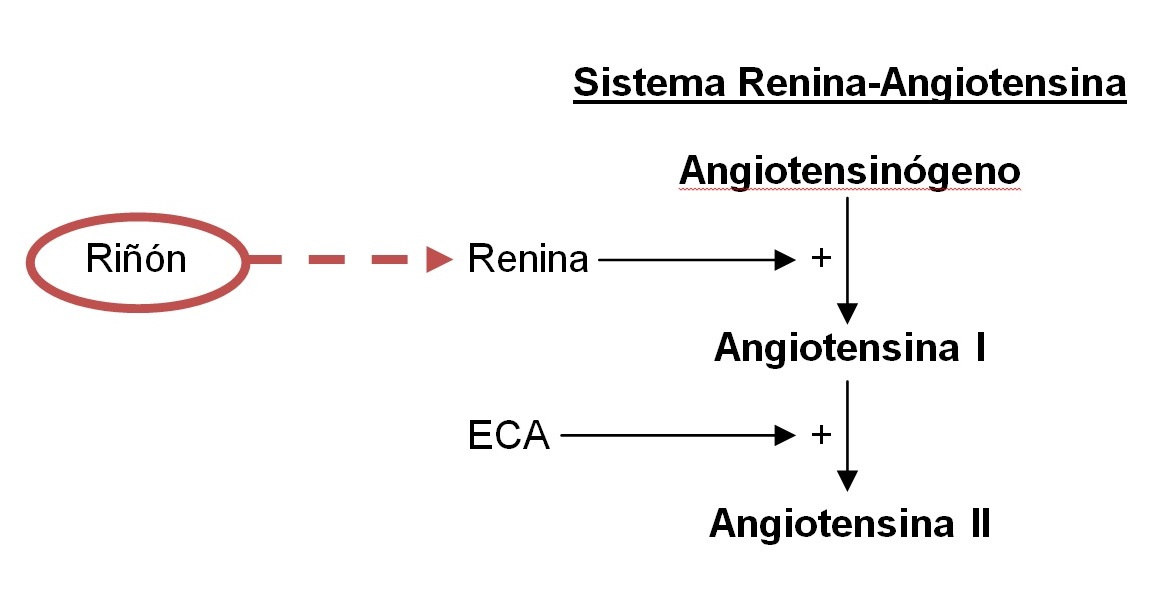 Hacia
la mitad de la década de 1950 se descubrió que la angiotensina
(hipertensina) en la sangre, un polipéptido que aumenta la presión
arterial, se produce en dos etapas: en la primera, una enzima originada
en el riñón, llamada renina, cataliza la liberación de un polipéptido
denominado la angiotensina I. La angiotensina I es un decapéptido
inactivo que carece de actividad presora pero la adquiere cuando otra
enzima diferente de la renina, presente en el plasma, libera el
dipéptido de la angiotensina I, y da lugar al octapéptido activo
denominado angiotensina II, con intensa acción presora (figura 1).
Leonard T. Skeggs y col. aislaron y purificaron esta enzima, que
denominaron Angiotensin Converting Enzyme (ACE) [Enzima de conversión de
Angiotensina (ECA)] y que resulta ser una metaloproteasa.
Hacia
la mitad de la década de 1950 se descubrió que la angiotensina
(hipertensina) en la sangre, un polipéptido que aumenta la presión
arterial, se produce en dos etapas: en la primera, una enzima originada
en el riñón, llamada renina, cataliza la liberación de un polipéptido
denominado la angiotensina I. La angiotensina I es un decapéptido
inactivo que carece de actividad presora pero la adquiere cuando otra
enzima diferente de la renina, presente en el plasma, libera el
dipéptido de la angiotensina I, y da lugar al octapéptido activo
denominado angiotensina II, con intensa acción presora (figura 1).
Leonard T. Skeggs y col. aislaron y purificaron esta enzima, que
denominaron Angiotensin Converting Enzyme (ACE) [Enzima de conversión de
Angiotensina (ECA)] y que resulta ser una metaloproteasa.El gran ímpetu en el comienzo de la investigación sobre los inhibidores de la enzima de conversión de angiotensina como agentes antihipertensores se debe al investigador brasileño Mauricio Rocha e Silva, el cual, en 1949, empezó a hacer pruebas con gotas de veneno extraídas de los colmillos de la Bothrops Jararaca Jaracussa, también conocida como la víbora brasileña del hoyo, reptil de color marrón oscuro, cuyo hábitat se encuentra entre las hojas de los árboles más famosos en la selva del Amazonas. Su veneno provoca la muerte de sus víctimas por una bajada de la presión arterial muy acusada. Mauricio Rocha e Silva descubrió, en el Instituto Biológico de São Paulo, que la inyección del veneno de la víbora en la circulación sanguínea de los mamíferos conduce a la producción de un importante péptido bioactivo hipotensivo y estimulante de la musculatura lisa denominada bradicinina. Este péptido está relacionado con el control de la presión sanguínea y con muchos otros procesos fisiológicos y patológicos.
Análogamente a lo que ocurre con la angiotensina I, la enzima de conversión de angiotensina es capaz de liberar el dipéptido de la bradicinina; pero, a diferencia de lo que sucede con aquella, este efecto da lugar a un producto que carece de las acciones farmacológicas de la bradicinina. La continuación de estos estudios en el laboratorio de Rocha e Silva, en la Univerdidad de São Paulo en Riberão Preto, permitieron que su alumno y colaborador, Dr. Sergio Ferreira1, descubriera, en 1965, que el veneno de la Brothrops jararaca contenía factores que potenciaban la acción del nonapéptido bradicinina. Tras fraccionar extractos del veneno consigue aislar estos factores (Bradiquinin Potentiating Factors, BPFs) que resultaron ser una familia de péptidos de 5 a 13 aminoácidos. Se demostró que su capacidad potenciadora de la bradicinina era consecuencia de su acción inhibidora de la degradación de la enzima. Un poco después, científicos del grupo del Dr Vane demostraron que los extractos crudos del veneno también inhibían la conversión enzimática de angiotensina I en angiotensina II2.
En los EE.UU., los investigadores de Squibb (Squibb Institute for Medical Research, Princeton, Nueva Jersey) Dres. Miguel Ondetti, David Cushmann y Bernard Rubin, estudiaban la enzima de conversión. Vane intentaba convencerles de que la inhibición de esta enzima ofrecería una nueva aproximación al control de la presión arterial. Sin embargo, los investigadores del Instituto Squibb no tenían claro que la potenciación de la bradicinina fuese una propiedad deseable en un antihipertensor debido a las propiedades proinflamatorias de este nonapéptido. Fraccionaron también los extractos del veneno de Bothrops jararaca pero valorando no la actividad potenciadora de bradicinina, sino la actividad inhibidora de la enzima de conversión. Resultó que ambas actividades residían en los mismos péptidos.
En 1971, Ondetti y sus colaboradores consiguen a partir del veneno identificar y sintetizar un péptido (el nonapéptido SQ 20.881), que resultó ser idéntico al nonapéptido BPPs (teprótido) el péptido más activo potenciador de bradicinina aislado por Ferreira.
Posteriormente, los estudios de Erwin G. Erdöss en la Universidad de Chicago con enzima de conversión homogénea mostraron que angiotensina I y bradicinina son sustratos para esta enzima. O, dicho de otro modo, la enzima responsable de la degradación de la bradicinina es también responsable de la formación de angiotensinaII.
La actividad dual de los péptidos de Ferreira (inhibidores de la degradación de bradicinina, un potente vasodilatador e inhibidores de la biosíntesis de angiotensina, un potente vasoconstrictor) los hace especialmente apropiados como prototipos para la investigación de agentes antihipertensores. El teprótido fue estudiado en animales y en el hombre. Descendía la presión arterial en pacientes con hipertensión esencial, pero su coste era muy elevado y su efectividad muy relativa, ya que solo era activo por vía parenteral, dado que los péptidos son destruidos durante la digestión. Se necesitaba un inhibidor de la enzima de conversión de angiotensina que fuese efectivo vía oral.
Como primer paso, imprescindible para ayudar a diseñar nuevos IECA, había que conseguir diseñar la estructura del receptor de la ECA y el hipotético punto de unión del inhibidor con la enzima3. Se observó que la actividad enzimática de la ECA es muy similar a la que presenta la carboxipeptidasa A, una enzima digestiva, teniendo ambos un punto de unión con su receptor conteniendo cinc. Esta observación ayudó a desarrollar un modelo hipotético de sitio activo de la enzima de conversión y, a partir de ahí, una nueva aproximación en la búsqueda de agentes antihipertensores.
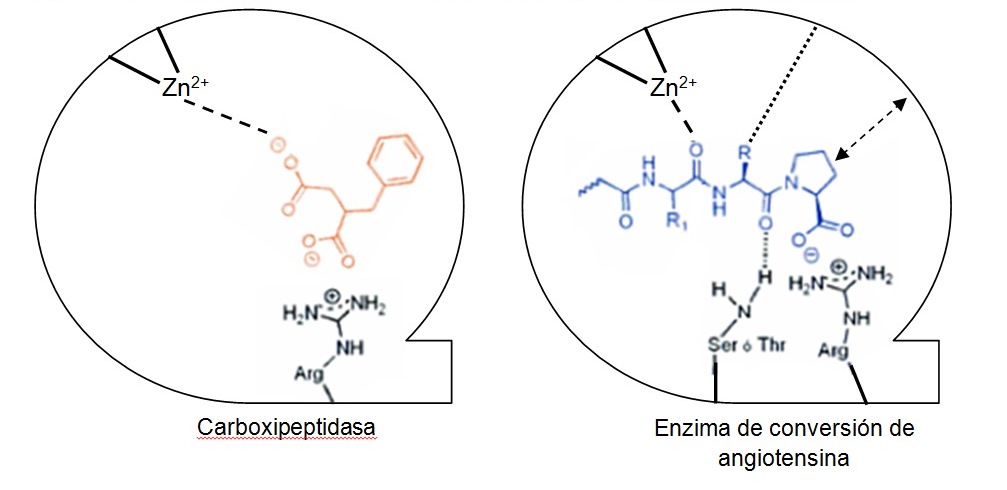 El desarrollo de los inhibidores de la enzima de conversión fue inspirado en la observación de Byers y Wolfenden4, que el ácido 2-bencilsuccínico es un potente inhibidor de la carboxipeptidasa A (figura 2).
El desarrollo de los inhibidores de la enzima de conversión fue inspirado en la observación de Byers y Wolfenden4, que el ácido 2-bencilsuccínico es un potente inhibidor de la carboxipeptidasa A (figura 2).
Los
investigadores de Squibb sabían que en la hidrólisis de la angiotensina
se liberaba no un aminoácido como en los sustratos de la
carboxipeptidasa, sino un dipéptido. Ello implicaba que la unión de un
inhibidor de la enzima de conversión que se uniese al sitio activo
tendría que ser de mayor longitud y pasaron a preparar péptidos con
residuos de alanil-prolina y otros residuos.
Cada posible candidato se valoraba in vitro, comprobando su actividad inhibidora sobre la ECA, empleando las propiedades contráctiles del íleon de cerdo guineano. Los ensayos estudiaron la contracción de la musculatura lisa, que produce un aumento de la permeabilidad de los vasos sanguíneos.
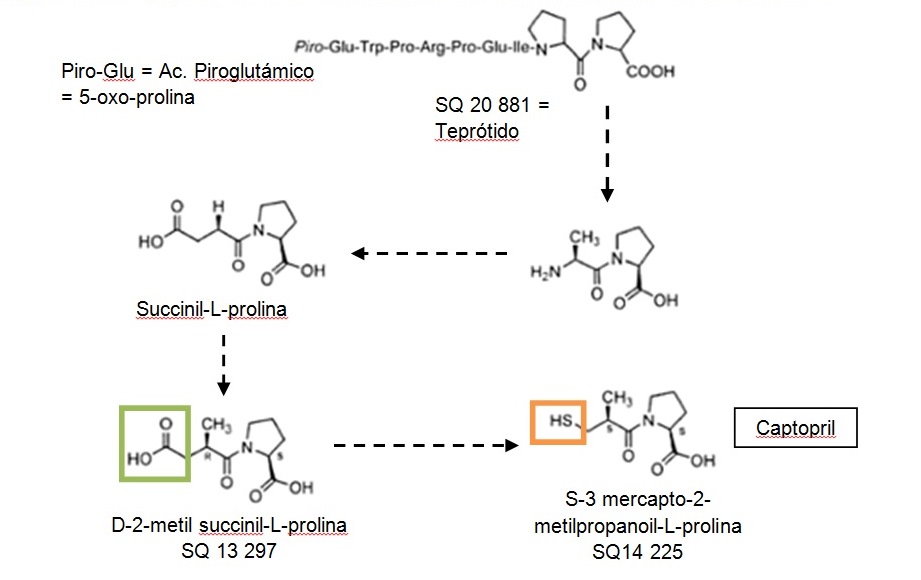
Encontraron que la succinil-L-prolina era un inhibidor, aunque débil, de la enzima de conversión. Este era un buen resultado, pero no lo suficiente. Por ese motivo, posteriormente, aplicando este concepto al modelo hipotético de ECA, Ondetti y col. prepararon series de derivados del ácido succínico con restos de prolina y de otros péptidos inhibidores existentes en los venenos de serpientes.
El primer inhibidor fue la succinil-L-prolina, al que le siguieron otros como D-2-metil-succinil-L-prolina o D-2-metil-glutaril-prolina. Ambos, derivados de la succinil-L-prolina, producen una actividad inhibitoria 14 veces mayor que la anterior y efectivos suministrados por vía oral.
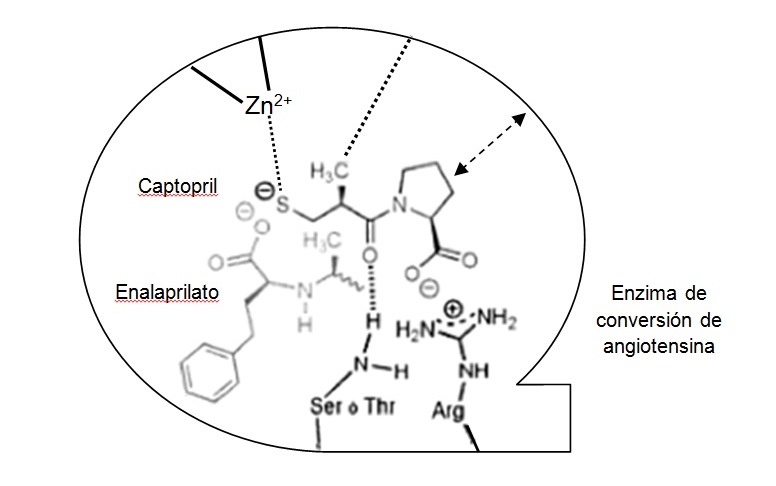
El paso siguiente consistió en sustituir el grupo carboxilo del extremo opuesto al que contiene el aminoácido por un grupo químico más adecuado para unirse con el Zn+, como es un grupo sulfhídrico (figuras 2 y 3). Así se obtuvo el captopril, el componente más activo logrado, 14.000 veces más que el succinil-L-prolina5.
 |
 |
La comunidad científica anuncia en 1977 la actividad antihipertensiva del captopril por vía oral6.
En 1981 fue aprobado como medicamento y estuvo disponible para el tratamiento de la hipertensión arterial7.
La investigación clínica indicó que era muy eficaz en el tratamiento de
la insuficiencia cardiaca, pues tenía una afinidad muy específica por
el sitio de unión de la ECA, con unos mínimos efectos secundarios.
Se había comprobado que el captopril era un medicamento de elección cuando los tratamientos convencionales antihipertensivos fallaban o mostraban demasiados efectos secundarios.
Los investigadores Ondetti y Cushman, por sus trabajos para la obtención del captopril, recibieron la concesión a la investigación médica de Albert Lasker, el equivalente americano del premio Nobel de medicina.
Históricamente, el descubrimiento del captopril es, a menudo, citado como el primer caso de diseño racional de un fármaco. En base al conocimiento de la estructura y función de una diana se diseñó una estructura para interaccionar con ella8.
Bibliografía
1. Ferreira, Sergio (February 1965). A bradykinin-potentiating factor (bpf) present in the venom of bothrops jararaca. British Journal of Pharmacology 24: 169-169.
2. Ng KKF and Vane JR: Fate of angiotensin I in the circulation. Nature, 1968, 218, 144-150.
3. Cushman D W and Cheung HS:Spectrophotometric assay and properties of the angiotensin-converting enzyme of rabbit lung. Biochemical Pharmacology 1971, 20, 1637.
4. Byers LD and Wolfenden R: Binding of the by-product analog benzylsuccinic acid by carboxypeptidase A. Biochemistry 1973, 12, 2070-2078.
5. DW Cushman, J Pluscec, NJ Williams, ER Weaver, EF Sabo, O Kocy, HS Cheung and MA Ondetti: Inhibition of angiotensin-converting enzyme by analogs of peptides from Bothrops jararaca venom. Experientia 1973, vol.29, 1031.
6. Ondetti MA., Rubin B., and Cushman DW: Design of specific inhibitors of angiotensin-converting enzyme: new class of orally active antihypertensive agents. Science 1977, 196, 441.
7. New Drug Application No. 18 343 Submitted by E. R. Squibb & Sons Inc. 1981, Approved April 6.
8. Enrique Raviña Rubira, Medicamentos Un viaje a lo largo de la evolución histórica del descubrimiento de fármacos II, Universidad de Santiago de Compostela, Servizo de Publicacións e Intercambio científico 2008, 625-635.
1. Ferreira, Sergio (February 1965). A bradykinin-potentiating factor (bpf) present in the venom of bothrops jararaca. British Journal of Pharmacology 24: 169-169.
2. Ng KKF and Vane JR: Fate of angiotensin I in the circulation. Nature, 1968, 218, 144-150.
3. Cushman D W and Cheung HS:Spectrophotometric assay and properties of the angiotensin-converting enzyme of rabbit lung. Biochemical Pharmacology 1971, 20, 1637.
4. Byers LD and Wolfenden R: Binding of the by-product analog benzylsuccinic acid by carboxypeptidase A. Biochemistry 1973, 12, 2070-2078.
5. DW Cushman, J Pluscec, NJ Williams, ER Weaver, EF Sabo, O Kocy, HS Cheung and MA Ondetti: Inhibition of angiotensin-converting enzyme by analogs of peptides from Bothrops jararaca venom. Experientia 1973, vol.29, 1031.
6. Ondetti MA., Rubin B., and Cushman DW: Design of specific inhibitors of angiotensin-converting enzyme: new class of orally active antihypertensive agents. Science 1977, 196, 441.
7. New Drug Application No. 18 343 Submitted by E. R. Squibb & Sons Inc. 1981, Approved April 6.
8. Enrique Raviña Rubira, Medicamentos Un viaje a lo largo de la evolución histórica del descubrimiento de fármacos II, Universidad de Santiago de Compostela, Servizo de Publicacións e Intercambio científico 2008, 625-635.
* 4.º Grado Farmacia. Farmacología. Facultad de Farmacia. Universidad CEU Cardenal Herrera. Tutora: Dra. Lucrecia Moreno Royo.
Fuente:
www.elfarmaceutico.es
Fuente:
www.elfarmaceutico.es







